Variant Comparison App (VCA) Workflow¶
Use cases¶
The variant comparison workflow allows for comparing datasets with similar contents. The overall workflow can be used to answer many different and generic sorts of question, but has specialized pieces that are specifically for comparing sequencing results from commercial vendors of tissue and plasma diagnostic assays.
Other questions that can be answered include:
- I'm running a technical replicate study and each sample has been sequenced/prepped/called by two different methods. What variants have been called in both?
- I have tissue and plasma samples from the same patient. Which variants are tissue-only? Plasma-only?
- I have plasma samples from the same patient taken at baseline and at variant timepoints during treatment. How did variant allele frequency vary during treatment?
- I want to know what genes are in common across multiple commercial diagnostic panels.
- I'm looking for a set of variants that are unique to my sample in this pooled set of samples.
The workflow is designed to allow any scientist to quickly create their own PlasmaSeq analysis1, from harmonizing many different sources of vendor data, creating merged datasets for comparison, and creating the same figures published in the paper.
Workflow¶
The overall workflow is composed of the following 4 steps/functions.
- Importing data in a variety of variant and variant formats
- Merging imported data into a single dataset
- Adding comparison fields to the merged dataset
- Analyzing the merged dataset
Import¶
Any dataset can be merged together that has entities in common. However, to take full advantage of the downstream visualizations and comparison features, the following fields need to be present.
| Field | Description | Example |
|---|---|---|
| sample | Sample ID must be the exact same string across all source datasets for the same sample. This may refer to a patient, a specific sample, or even a timepoint, depending on what you are comparing. | TCGA-02-0001 |
| variant | Must be in SolveBio's variant ID format | GRCH37-7-55249071-55249071-T |
| allelefreq | Allele frequency of the variant. Also known as allele fraction or percentage of reads. | 0.021 |
Your sample IDs MUST match
Your sample IDs must be EXACTLY the same across all source datasets. You may need to perform remapping expressions on your source datasets to get the right sample IDs in.
The following diagnostic report output formats are supported as part of SolveBio's template system and already include the fields described above. When data is imported using any of the following templates, it will automatically be ready for the VCA workflow.
- Vardict
- Foundation Medicine OAPL
- Guardant Health
- Resolution Bio
To import a dataset with a template, select the relevant text file on SolveBio, and then import with the correct template.
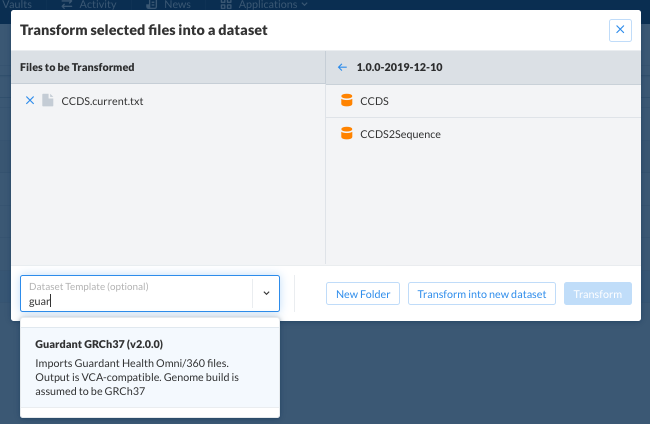
Merging¶
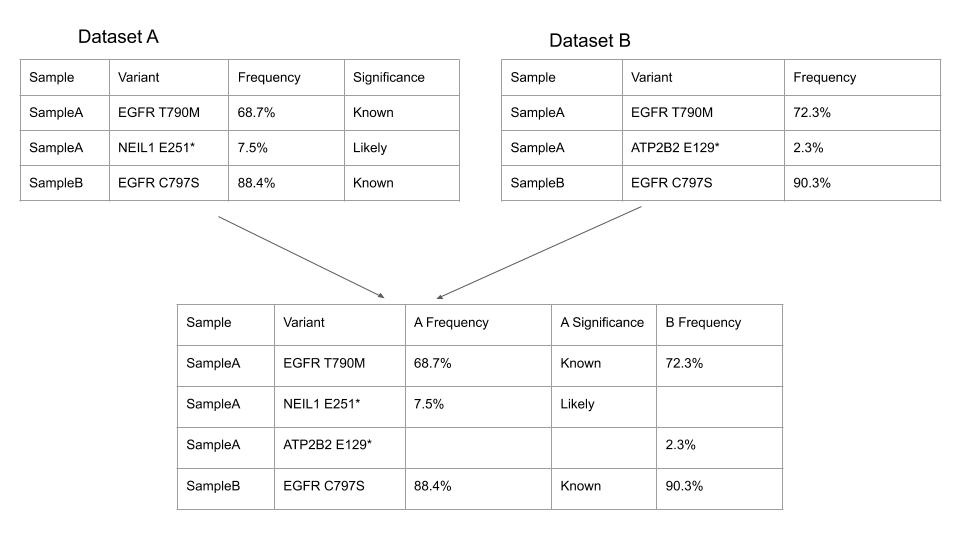
Merging of datasets on SolveBio is conceptually simple to a pandas DataFrame outer merge or a SQL full outer join.
Select the datasets to be merged by adding as many datasets as necessary to the merge in the Vault view.
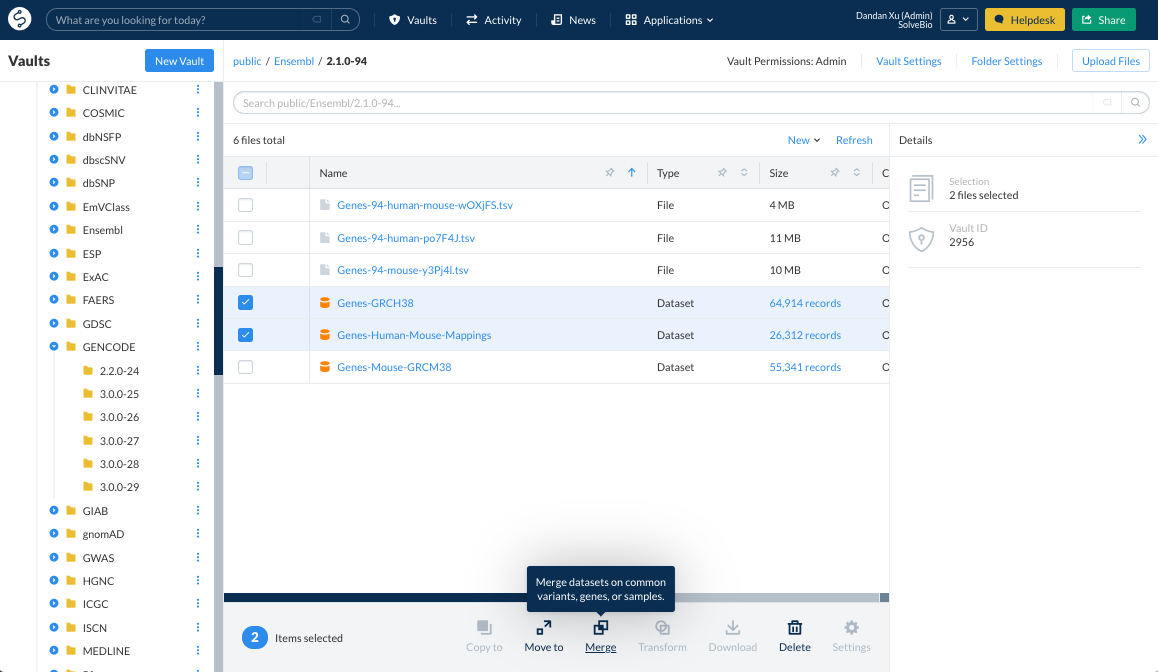
Each source dataset gets a user-defined label. Saved queries can also be used to reduce the amount of data being merged.
Select which entities to use as "merge keys". These entities together become the common unique identifier across all datasets in the merge and is the unique ID for the record in the new merged dataset. For example, if sample and variant are selected as the merge keys, then the final merged dataset consists of all the unique sample-variant value combinations in the source datasets that were merged into the final dataset. Typically, sample and variant are the correct entities to use.
Optionally, select fields from each source dataset to be included in the final dataset. This is useful for comparing variant classifications from different sources (e.g. vardict's significance versus Foundation Medicine's somatic status), different gene/transcript annotations, variant allele frequencies across different samples, etc. In the merge workflow on SolveBio, commonly used fields from the supported import templates are auto-selected for convenience.
Comparison Analysis¶
The comparison part of the VCA workflow computes true positives/negatives and false positives/negatives using truth set(s) or by consensus (in 2 or more datasets for the same sample-variant pair). Optionally select a sequencing panel to determine detectability of each variant by panel. Comparison analysis can only be turned on where sample-variant are selected as the merge-key entities.
If comparison analysis is turned on, an additional filter is added in the background where only samples that are present in all source datasets are included into the merge. If NO sample is present in all source datasets, then this filter is not added.
During the setup of the merge, there is an option to add "comparison" analysis fields to the final dataset. These comparison fields are added after the merged dataset is created from all the source datasets, in a second set of annotations.
| Field | Description | Example |
|---|---|---|
| found_in | Label of the datasets this merge key combination was found in | Baseline |
| n_datasets | Number of datasets this merge key combination was found in | 3 |
| detectable | List of panels that this variant overlaps, if there is a variant in this dataset and panels have been given as input. | FMI CDX |
| call | Ground truth call for this record. A record (merge-key combination, e.g. a variant within this sample) can be considered true (or "real") if it was found in a truth dataset or was considered truth through consensus (found in at least 2 datasets). | True |
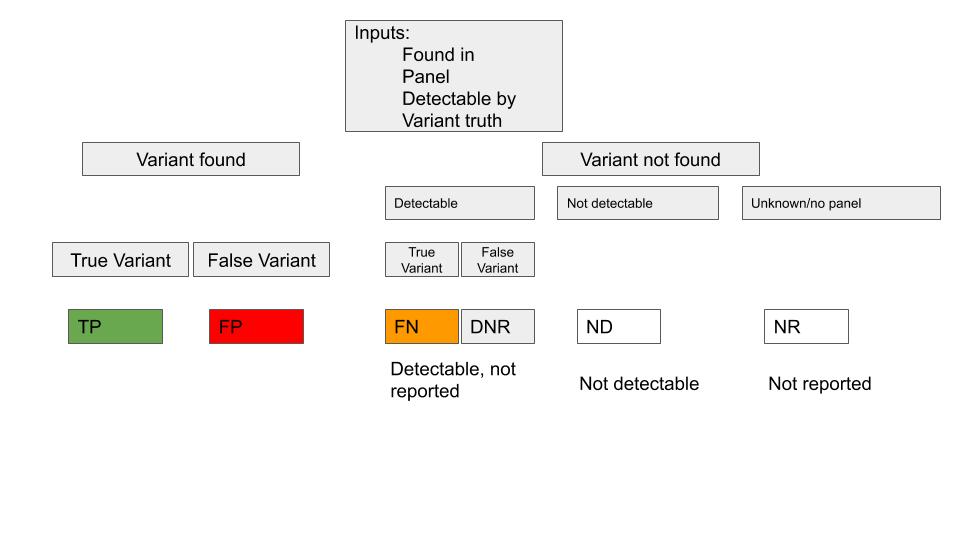
| calls | An dictionary that contains how each source dataset called the variant. Based on whether or not the variant has been deemed a true or false variant within the call field. | TP |
The definition for the possible values of calls are: TP = true positive (should have found it, was found) FN = false negative (should have found it, not found) FP = false positive (should not be found, was found) DNR = detectable, not reported (could have found it, did not) ND = not detectable (could not have found it, did not) NR = not reported (no panel information was given)
Concordance logic
Finally, the merged dataset is created and tagged with the tag VCA. Depending on the size of the source datasets and the amount of data to be merged, this dataset may take a while to finish all activity (datasets over 10 million records should be filtered down for best results).
Analysis¶
After the merged dataset has finished importing all the source datasets and the comparison annotations are complete, the dataset is ready for analysis. Analysis can be as simple as using the SolveBio data table view to filter by which variants are detectable, false positives, etc, adding additional annotations using SolveBio Expressions, or exporting the data to sort and filter in Excel and visualize in Spotfire.
The dataset may also be opened in the Comparison Report. This report only displays 250 variants, so for best results, filter down the dataset to <250 variants and then click Open in > Comparison Report. This visualization app displays 3 figures:
1) Variant concordance plot - scatter plot of allele frequencies categorized by source dataset and classification
2) Direct comparison plot - scatter plot of allele frequencies categorized by source dataset, with lines between source datasets where the variant exists in multiple
3) Patient-level tile plot - Same tileplot as in the PlasmaSeq paper, displays individual variants, their call status (TP/FP/etc), and allele frequencies from each source dataset
Only the first 250 variants will be visualized
The comparison has a strict 250 variants limit! Only the frist 250 will be visualized, the rest will be ignored. Filter the dataset to below 250.
Additional help¶
SolveBio regularly performs advanced comparison setups and analysis for customized comparisons. Please contact support@solvebio.com for more information.
-
Orthogonal Comparison of Four Plasma NGS Tests With Tumor Suggests Technical Factors are a Major Source of Assay Discordance Daniel Stetson, Ambar Ahmed, Xing Xu, Barrett R.B. Nuttall, Tristan J. Lubinski, Justin H. Johnson, J. Carl Barrett, and Brian A. Dougherty JCO Precision Oncology 2019 :3, 1-9 https://ascopubs.org/doi/full/10.1200/PO.18.00191 ↩